










関連記事



ChIP-seqトラブルシューティング101:低信号のステップバイステップ修正
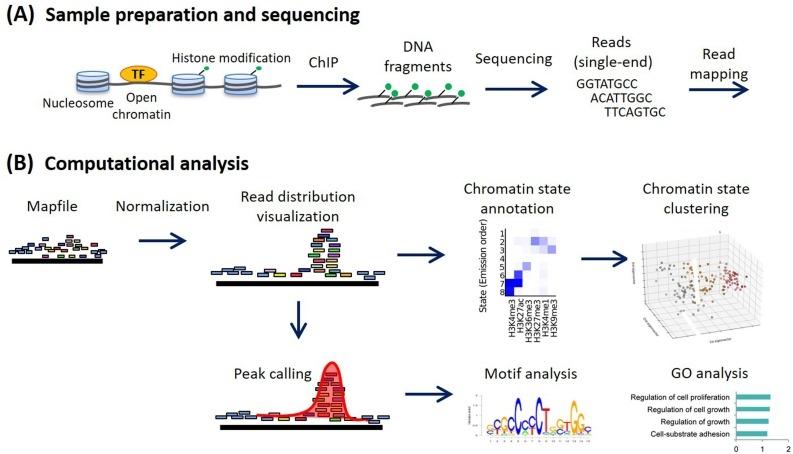
2026-02-28ChIP-Seqトラブルシューティングは、しばしば単純な症状である低信号から始まりますが、実際の原因は通常、クロマチン質、抗体性能、免疫沈殿条件、ライブラリーQCに分散しています。Longlight Technologyでは、ヒストンマークや転写因子のChIP-Seqを実施するラボを支援しており、同じ「静かな故障点」が異なる器具やサンプルタイプで繰り返されています。このChIP-Seqトラブルシューティング入門ガイドは、実用的なチェックポイントを用いて、一度の実行で検証できるものを使い、論理的な順序で信号を回復するためのステップバイステップを初心者に案内します。
ChIP-seq解析の手法:実践的なワークフローと高度な応用 - ScienceDirect
1) 定義 "信号が低い" 何かを変える前に
信号が少ないことは、ピークが少なすぎる、既知の座での濃縮が弱い、あるいはライブラリーが問題なく見えるがマッピングが悪いなど、さまざまな意味を意味します。 良いChIP-Seqトラブルシューティング まずは故障モードを明確にラベル付けすることから始めます。なぜなら、各モードは異なる修正を指しているからです。
初心者にわかりやすい定義方法は、3つの層を確認することです。
✓ 生物学層:ターゲットは細胞状態(刺激状態か安静状態か、分化段階、治療時期)に存在しますか?
✓ 濃縮層:ChIPのDNAはqPCRによる濃縮が陽性コントロール遺伝子座で示されるのか、それとも陰性領域で見られるのか?
✓ シーケンシングレイヤー:十分な一意でマッピングされたリードと適切な重複レベルがありますか?
これら3つに答えられないなら、「すべてを最適化する」のはやめましょう。まずは1つのコントロール実験を行いましょう:サンプル数は同じに保ち、シーケンス深度は控えめにし、疑われる変数は1つだけ変更します。
2) クロマチンから始める:断片サイズ あるND損失制御
検査室で「なぜChIPが機能しないのか」と尋ねられると、最も一般的な根本原因はクロマチンが過剰に断片化されている、断片化不足、あるいはクリーンアップ時に単に失われていることです。ChIP-Seq トラブルシューティングでは、クロマチンが基盤となります。もし不安定であれば、その後のステップはノイズになります。
超音波処理ベースのワークフローでは、多くの研究室が鋭いピークコールと安定した免疫沈降のために~150〜300 bpの範囲の断片を目標としています。断片が主に大きい場合(例えば>500 bp)、抗体は標的複合体を効率的に引き落とすのに苦労します。断片が小さすぎると、非特異的なDNAを放出してエピトープを破壊したり、背景を増やしたりすることがあります。
すぐに実行できる実践的なチェックポイント:
• 逆架橋およびクリーンアップ後のDNA測定(IP前だけでなく)。ここで大きく下がると、ビーズやカラム、または過酷な環境での損失を示します。
• サンプル間の断片化プロファイルを比較する。あるサンプルが「完璧」で、次のサンプルがにじみや過剰サイズの場合は、抗体ではなく溶解と超音波検査の設定に注目してください。
• 各バッチから「入力DNA」を保持すること。これがqPCRとライブラリQCの両方の基準です。
Longlight Technologyでは、クロマチン準備を管理された製造工程として扱うことを推奨しています。バッファー組成を固定し、超音波検査中のサンプル温度を安定させ、正確なサイクル設定を記録します。わずかな偏差が後でピーク強度に大きな差を生みます。
3) 抗体適合性:特異性、ロットバリア、 あるNDコントロール
クロマチンが妥当に見える場合、ChIP-Seqトラブルシューティングの次のステップは抗体の選択と検証です。信号が低いのは、しばしば「ウエスタンには良い」抗体がチチップには弱い抗体を使うこと、またはロットごとの変動性によって引き起こされます。
良い抗体戦略は以下のコントロールを中心に構築されます:
✓ ポジティブコントロールターゲット:ロバースエンリッチメントを持つヒストンマーク(ワークフローの健全性確認に一般的に用いられます)。
✓ 陰性対照:IgGまたはアイソタイプコントロールで背景プルダウンを推定します。
✓ 既知の遺伝子座qPCR:ターゲットに対して1つまたは2つの陽性遺伝子座が発表され、さらに遺伝子デザート領域が1つあります。
転写因子の場合、シグナルはヒストンマークよりも本質的に低い場合があります。つまり、抗体親和性が高く、IP条件がクリーンでなければなりません。TF ChIPが初めての方は、シーケンスの深さを変えることから始めないでください。まずqPCRで濃縮を確認しましょう。qPCR濃縮が弱い場合、リードを多く行うとノイズが増えます。
実用的なコツ:抗体ロットを切り替える際は、可能であれば同じクロマチンバッチでの濃縮を再度確認してください。ロットチェンジが信号を壊すなら、ワークフロー自体が必ずしも「間違っている」わけではありません。試薬の性能が変わったのですし、トラブルシューティングの経路もそれを反映しているはずです。
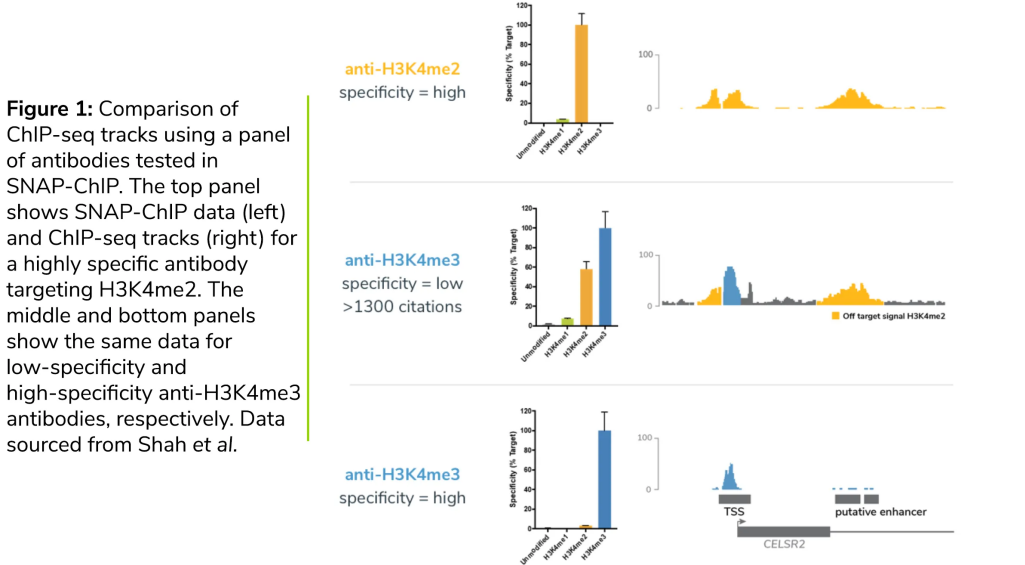
実験に適したChIP抗体の選び方 - EpiCypher
4) クリーンなIP最適化シーケンス—推測なし
クロマチンや抗体を超えて、IP化学(ビーズ、ウォッシュ、インキュベーション)が次のホットスポットです。信号が低いのはしばしば背景の問題です。
✓ ビーズ選択:抗体種やアイソタイプに合ったプロテインA/Gを選びます。
✓ 抗体量:弱いプルダウンや非特異的DNAの上昇を避けるために滴定。
✓ ウォッシュバランス:ノイズを除去しつつ、弱いが実際の相互作用を保持するために厳密さを調整します。
実用的な初心者のルールとして、1回のランで軸を1つだけ調整することです:
・ピークが存在するが弱い場合は、有効捕捉を増加させる(抗体がわずかに増え、ビーズ結合が改善され、培養期間が長くなる)。
・ピークが広くノイズが多い場合は特異性を高め(ウォッシュの強化、ブロッキングの改善、抗体過負荷の軽減)。
メーカーの視点から見ると、ロングライト・テクノロジーは取り扱い時の損失を最小限に抑える免疫沈殿試薬と磁気ビーズシステムを設計しています。なぜなら、サンプル損失はまさに「低信号」のように見えるからです。滑らかなビーズ分離、一貫したバインディング、清潔な洗浄工程により、オペレーター間のばらつきが減り、特に新人を育成するチームにとって重要です。
5) ライブラリの品質管理:いつ "良いDNAです" それでも信号は低いままです
時にはChIPのDNA濃縮が実際に行われることもありますが、最終的なデータは依然として平坦に見えます。ChIP-Seqトラブルシューティングでは、通常ライブラリ構築やシーケンスメトリクスを指しています。
信号低下の一般的なライブラリレベルの原因:
✓ 過剰増幅:PCRサイクルが多すぎると重複が増え、利用可能なユニークリードが減少します。
✓ アダプター/プライマーアーティファクト:これらはシーケンシングリードを消費し、ターゲットカバレッジの向上を妨げます。
✓ 複雑性の低さ:多くの場合、非常に低いChIPのDNA入力やクリーンアップ中の損失が原因です。
ChiIP全体を再実行する前に確認すべきこと:
・ライブラリサイズ分布(複数の予期せぬピークではなく、クリーンなピークが望ましい)。
• アライメント後の重複率および一意マッピング率。
• ピークのリード数(FRiP)の傾向は内部ベースラインと比較的(初心者でも「良いか悪いか」をランごとに追跡できる)。
ライブラリーのオーバーサイクリングが疑われる場合は、サイクル数を減らし、上流のキャプチャ効率を向上させる(クロマチン回収率の向上とIP特異性の向上)という簡単な改善策があります。より深いシーケンスでは、低複雑度ライブラリを補うことはできません。
6) ステップ-著者-ステップ信号の回復は明日から始められます
アドホック調整を上回る実用的なシーケンス:
✓ ステップ1:クロマチン断片が~150–300 bpであることを確認し、逆交差後のDNA回復を確認します。
✓ ステップ2:ライブラリ準備前にqPCRによる濃縮を1つの陽性遺伝子座と1つの陰性遺伝子座で確認します。
✓ ステップ3:「無濃縮」と「高背景」を区別するために適切なコントロール(入力IgG)を追加。
✓ ステップ4:IP条件を1変数ずつ調整します(ビーズ、抗体量、洗浄の厳しさ)。
✓ ステップ5:シーケンス深さが問題であると仮定する前に、ライブラリの指標(重複、マッピング、サイズ分布)を監査します。
CTA(ロングライト・テクノロジー): より速い方法を望むなら、Longlight Technologyに連絡してChIP-Seqトラブルシューティングのチェックリストとサンプルごとの診断ワークシート(クロマチン→IP→ライブラリ)を入手してください。また、操作者の変動を減らし、初心者がより早く安定した濃縮に達するのを支援するために、対照設計や試薬の組み合わせ戦略も推奨できます。
信号が低いのはイライラしますが、めったに謎めいたものではありません。クロマチンの完全性、抗体適合性、IP特異性、そして最終的にライブラリの品質管理から始まる規律あるChIP-Seqトラブルシューティングのフローを用いれば、1回の弱い実行をサンプル、スタッフ、プロジェクトにスケールする再現可能なプロトコルに変えることができます。